









содржина
Бурневилска туберозна склероза
Што е тоа ?
Бурневилската туберозна склероза е сложена генетска болест која се карактеризира со развој на бениген (неканцероген) тумор во различни делови од телото. Овие тумори потоа може да се лоцираат во кожата, мозокот, бубрезите и другите органи и ткива. Оваа патологија може да предизвика и сериозни проблеми во развојот на поединецот. Сепак, клиничките манифестации и тежината на болеста варираат од пациент до пациент.
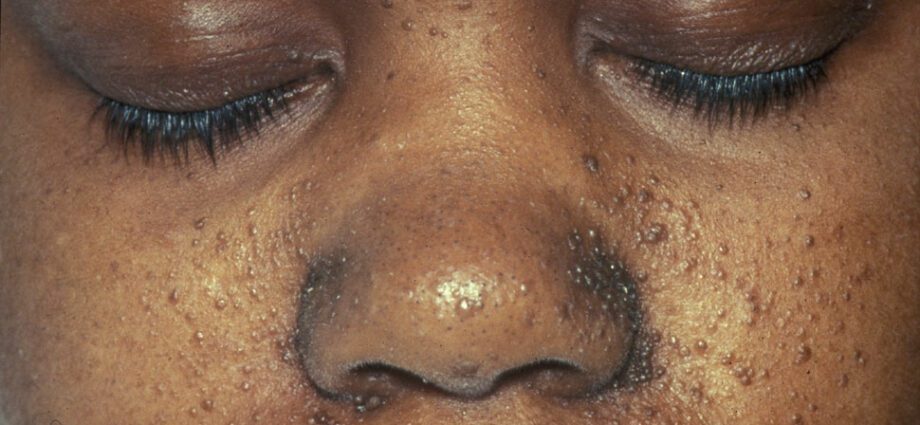
Придружните абнормалности на кожата се генерално слични на дамки на кожата или на области каде што кожата е посветла отколку на остатокот од телото. Развојот на тумори на лицето се нарекува ангиофибром.
Во контекст на оштетување на мозокот, клиничките знаци се епилептични напади, проблеми во однесувањето (хиперактивност, агресивност, интелектуална попреченост, проблеми со учењето итн.). Некои деца со оваа болест имаат дури и некаква форма на аутизам, развојни нарушувања, кои влијаат на социјалните интеракции и комуникацијата. Бенигните тумори на мозокот исто така може да предизвикаат компликации кои можат да бидат фатални за субјектот.
Развојот на тумори во бубрезите е вообичаен кај луѓето со туберозна склероза. Ова може да предизвика сериозни компликации во функцијата на бубрезите. Покрај тоа, тумори може да се развијат во срцето, белите дробови и мрежницата. (2)
Тоа е ретка болест, чија преваленца (број на случаи во дадена популација во дадено време) изнесува од 1/8 до 000/1 луѓе. (15)
симптомите
Клиничките манифестации поврзани со туберозна склероза на Бурневил варираат во зависност од засегнатите органи. Покрај тоа, симптомите поврзани со болеста варираат во голема мера од една до друга личност. Со симптоми кои се движат од благи до тешки.
Најшироко идентификувани симптоми на оваа болест вклучуваат епилептични напади, когнитивни и бихејвиорални нарушувања, абнормалности на кожата итн. Органите кои најчесто се засегнати се: мозокот, срцето, бубрезите, белите дробови и кожата.
Развојот на малигни (канцерогени) тумори е можен кај оваа болест, но се ретки и главно ги зафаќаат бубрезите.
Клиничките знаци на болеста во мозокот потекнуваат од напади на различни нивоа:
– оштетување на кортикалните туберкули;
– епендимални нодули (SEN);
– џиновски епендимални астроцитоми.
Тие резултираат со: развој на ментална ретардација, тешкотии во учењето, нарушувања во однесувањето, агресивност, нарушувања на вниманието, хиперактивност, опсесивно-компулсивни нарушувања итн.
Оштетувањето на бубрезите се карактеризира со развој на цисти или ангиомиолипоми. Тие можат да доведат до бубрежна болка, па дури и до откажување на бубрезите. Ако е забележливо обилно крварење, тоа може да биде од тешка анемија или висок крвен притисок. Може да бидат видливи и други посериозни, но ретки последици, особено развој на карциноми (тумор на составните клетки на епителот).
Оштетувањето на очите може да биде слично на видливи точки на мрежницата, предизвикувајќи визуелни нарушувања или дури и слепило.
Абнормалности на кожата се многубројни:
– хипомелански макули: кои резултираат со појава на светли дамки на кожата, каде било на телото, како резултат на недостаток на меланин, протеин кој и дава боја на кожата;
– појава на црвени дамки на лицето;
– обезбојани дамки на челото;
– други абнормалности на кожата, зависни од една до друга личност.
Лезиите на белите дробови се присутни кај 1/3 од пациентите со мала доминација на жените. Придружните симптоми потоа се повеќе или помалку сериозни тешкотии со дишењето.
Потеклото на болеста
Потеклото на болеста е генетско и наследна.
Преносот вклучува мутации во гените TSC1 и TSC2. Овие гени од интерес влегуваат во игра во формирањето на протеините: хамартин и туберин. Овие два протеини овозможуваат, преку интерактивна игра, да се регулира клеточната пролиферација.
Пациентите со оваа болест се раѓаат со најмалку една мутирана копија од овие гени во секоја нивна клетка. Овие мутации потоа го ограничуваат формирањето на хамартин или тубертин.
Во контекст каде што двете копии на генот се мутирани, тие целосно го спречуваат производството на овие два протеини. Затоа, овој недостаток на протеини повеќе не му дозволува на телото да го регулира растот на одредени клетки и, во оваа смисла, доведува до развој на туморски клетки во различни ткива и/или органи.
Фактори на ризик
Факторите на ризик за развој на таква патологија се генетски.
Навистина, преносот на болеста е ефикасен преку автосомно доминантен начин. Или, мутираниот ген од интерес се наоѓа на несексуален хромозом. Дополнително, присуството на само една од двете копии на мутираниот ген е доволно за болеста да се развие.
Во оваа смисла, поединецот кој поседува еден од овие двајца родители кој страда од оваа болест има 50% ризик да го развие самиот болен фенотип.
Превенција и третман
Дијагнозата на болеста е пред сè диференцијална. Се заснова на атипични физички критериуми. Во повеќето случаи, првите карактеристични знаци на болеста се: присуство на повторливи епилептични напади и доцнење во развојот на субјектот. Во други случаи, овие први знаци резултираат со дамки на кожата или идентификација на тумор на срцето.
По оваа прва дијагноза, дополнителни испитувања се неопходни за да се потврди дијагнозата или не. Тие вклучуваат:
– скенирање на мозокот;
– МРИ (магнетна резонанца) на мозокот;
– ултразвук на срцето, црниот дроб и бубрезите.
Дијагнозата може да биде ефикасна при раѓањето на детето. Во спротивно, важно е тоа да се спроведе што е можно побрзо за да се преземе одговорноста за пациентот што е можно поскоро.
Во моментов, не постои лек за болеста. Затоа, поврзаните третмани се независни од симптомите што ги прикажува секој поединец.
Обично, се даваат антиепилептични лекови за да се ограничат нападите. Покрај тоа, се препишуваат и лекови за третман на туморски клетки на мозокот и бубрезите. Во контекст на проблеми во однесувањето, неопходен е специфичен третман на детето.
Третманот на болеста обично е долгорочен. (1)